Ученые Института химической биологии и
фундаментальной медицины СО РАН совместно с коллегами из Швеции и Москвы
научились определять подвижную структуру белка. Они исследовали сериновую
протеазу вируса Денге. Знания о структуре помогут создать противовирусные
препараты. Исследование опубликовано в журнале Nature Communications
Biology.
Специалисты изучали сериновую протеазу вируса
Денге. Он распространяется из Африки и Азии в страны Европы и передается людям
от комаров. В России есть потенциальная угроза возникновения очагов заболевания
в южных регионах. При распространении инфекции в организме человека
синтезируется длинная полипептидная цепочка. Протеаза разрезает эти цепочки на
более короткие фрагменты, которые становятся активными белками вируса, из них
формируется структура новых его копий. Если получится нарушить работу протеазы,
то вирус перестанет размножаться в организме человека. Кроме того, ингибитор
для Денге поможет создать такой же препарат для вируса клещевого энцефалита,
ведь обе инфекции относятся к одному семейству.
«Обычно, когда создают лекарство, берут
пространственную структуру белка и смотрят, как к ней присоединить какую-либо
молекулу, которая бы мешала работе активного центра белка и не давала вирусу
нормально функционировать. Чаще всего белок имеет довольно стабильную структуру.
В нашем же случае она очень подвижна, поэтому возникают проблемы», — рассказал
заведующий лабораторией структурной биологии, заместитель директора по научной
работе ИХБФМ СО РАН кандидат физико-математических наук Александр Анатольевич
Ломзов.
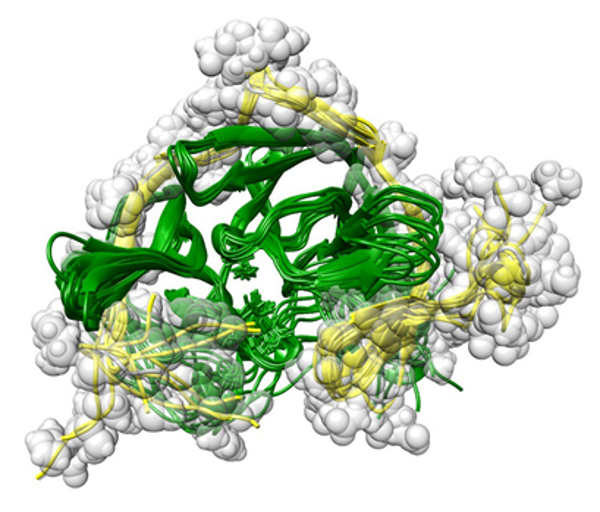
Структура комплекса белков сериновой протеазы
вируса Денге определенная в ходе исследований. Желтым цветом обозначен остов,
серым — атомы белка NS2B; зеленым цветом показан остов белка NS3pro
Еще одна сложность заключается в том, что
пространственная структура сериновой протеазы вируса Денге может изменяться при
переходе от неактивной формы к активной. В результате количество вариантов
структур, которые необходимо изучить, становится просто огромным. Поэтому
ученые задались вопросом, для какой из структур необходимо искать ингибитор, и
начали разрабатывать методику, чтобы найти нужную. Они скомбинировали классические,
уже известные подходы и предложили новый метод. К исследованию подключилась и
команда Института биоорганической химии им. академиков М. М. Шемякина и Ю. А.
Овчинникова РАН, которая разрабатывает методы для расчета и определения
динамических параметров структур белков.
«Один из подходов (метод ядерного магнитного
резонанса) позволяет измерять расстояние между отдельными атомами. Мы получаем
набор парных расстояний и на основе этих данных с использованием методов
компьютерного моделирования восстанавливаем структуру белка. Однако для таких
динамических систем одному и тому же набору парных расстояний соответствуют
сразу несколько структур. Какую из них выбрать? Нужно было проверить,
действительно ли они удовлетворяют другим критериям, выбрать эти критерии и
соотнести с экспериментом, потому что просто по расчетным характеристикам
подходят все. Мы разработали методику, чтобы искать набор интересующих нас
структур. Получилась такая схема: сначала синтезируем белок, исследуем его
методами ЯМР. Дальше на основе этих данных проводим компьютерное моделирование,
получаем набор структур. Снова проводим компьютерное моделирование, но более
детальное, рассчитываем динамические параметры, смотрим, как двигаются и
взаимодействуют отдельные атомы. После все эти данные мы объединяем и
анализируем в совокупности, делаем вывод о том, какие структуры могут
присутствовать в растворе», — отметил Александр Ломзов.
Структуру белка необходимо исследовать в
растворе чтобы быть максимально приближенным к биологическим условиям. Еще одна
сложность исследования в том, что этот белок, сериновая протеаза, начинает
расщеплять сам себя. Поэтому для его изучения необходимо либо вводить мутации в
структуру и делать белок неактивным, либо использовать вещества, которые
снижают его активность, либо сразу же после получения исследовать. Поэтому все
эксперименты проводят в растворе.
Ученые проанализировали три модельные структуры
и показали, что все они могут существовать, подтвердили предположение о том,
что белки динамичны и набор конформаций может быть разным.
«Это важно учитывать при поиске терапевтических
соединений. Сейчас у нас уже есть модельное соединение, которое
предположительно может сработать. Мы исследуем, как оно взаимодействует с
белком, где находится, какая структура у этого комплекса. Знания о структуре
необходимы, чтобы понять в деталях, как функционирует белок. Дальше их можно
использовать для создания и улучшения ингибитора», — сказал Александр Ломзов.
Ученый отметил, что провести такое обширное
исследование удалось благодаря работе молодежной лаборатории: «Для нашей
молодежной лаборатории мы построили вычислительный комплекс. Если бы его не
было, мы бы существенно медленнее двигались к получению результатов».